
He knows the evolution of Huntington's disease and how to treat
Written by:Huntington's disease (HD) is a rare movement disorder. It is hereditary and is characterized by involuntary movements (chorea), psychiatric disorders and cognitive impairment.
Causes of Huntington's disease
The cause of Huntington's disease is a defect in a gene (IT15), encoding a protein called huntingtin. The genetic defect results in the production of an abnormally long protein (mutant huntingtin), which would be responsible for neuronal degeneration that occurs in the disease. In the brain there is neuronal loss and huntingtin deposits and other proteins in the form of nuclear and cytoplasmic inclusions in surviving neurons.
There is no relationship between Huntington and Parkinson's disease, although in both a process of neurodegeneration and deposition of abnormal proteins in the brain occurs: hungtintina in HD and synuclein in Parkinson's. In Parkinson's, unlike what happens in HD, the patient's movements are characteristically slow down. Huntington's disease there is excess mobility (hyperkinesia).
There is also an important clinical exposure between HD and other neurological hereditary diseases.
When and how Huntington's disease appears
The average age of onset of symptoms is 38 years with variable boundaries between the second and seventh decades of life. The clinical picture set consists of three cardinal manifestations: choreic hyperkinesia, neuropsychiatric disorders and cognitive impairment. Chorea affects the muscles but also axial extremities. Also, there dysarthria and sometimes dysphagia as a result of a dysfunction of the pharyngeal muscles and upper oesophageal. The framework of neuropsychiatric disorders is very variable, from subtle personality changes and depression to severe psychotic disorders. There is a high incidence of suicide in families with Huntington's disease.
Differential diagnosis of Huntington's disease
Diagnosis is based on clinical data and checking vertical transmission of autosomal dominant inheritance. Neuroimaging (CT and MRI) show typical alterations. Confirmation of EH, and presymptomatic diagnosis can be made by molecular genetic techniques that demonstrate the pathological CAG triplet expansion in the IT15 gene.
We know that between 1 and 7% of patients clinically diagnosed EH do not have a gene mutation IT15. These subjects with a clinical diagnosis of Huntington 's disease but IT15 negative, has come to be called Huntington disease them-like (HDL).
In addition to genetic causes, there are many Koreas acquired to be considered in the differential diagnosis of chorea in HD. These include Sydenham's chorea, Wilson's disease, hyperthyroidism, the choreic drug-induced dyskinesia (tardive dyskinesia) disease or Gilles de la Tourette.
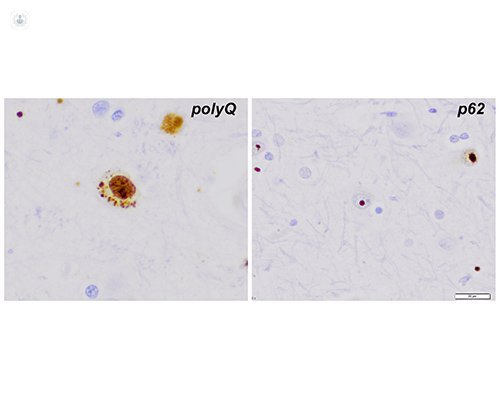
Histology also shows the significant loss of medium spiny neurons in the basal ganglia, the intranuclear accumulation of CAG repeats, as seen by immunohistochemistry with antibodies against polyglutamine expansions (left panel, polyQ). Compact intranuclear inclusions are visible using anti - ubiquitin or anti - p62 immunostaining (right panel, p62)
Forecast Huntington's disease
Huntington's disease invariably follows a progressive clinical course that is faster in juvenile-onset forms. The average duration of HD is 19 years, with limits between 10 and 25 years.
Treatment of Huntington's disease
The treatment can alleviate some demonstrations, but does not delay or onset or progression of symptoms. Treatment should be multidisciplinary and should support not only the patient but also their families, so it is advisable to intervene, besides experts in neurology , social workers, psychologists and other professionals familiar with the problems of these patients.
The only drugs that have proven effective in controlling chorea include antagonists of dopamine receptors in the striatum (neuroleptics) inhibitors storage or release of dopamine (reserpine and tetrabenazine). There is no effective treatment for dementia, the most debilitating aspect of the disease, but antipsychotic and antidepressant drugs are useful in the therapy of other neuropsychiatric manifestations.
In addition, Huntington's disease is essential to appropriate genetic counseling. Detecting carriers of the disease, presymptomatic stages, is possible thanks to new techniques of molecular biology. Since the application of these is not without legal and ethical problems in many hospitals have set up committees in participating patients, doctors, lawyers and medical ethicists, in order to assess, in each case, on whether to implement them.

