
Egli conosce l'evoluzione della malattia di Huntington e come trattarla
Autore:La malattia di Huntington (HD) è un disturbo del movimento rara. E 'ereditaria ed è caratterizzata da movimenti involontari (corea), disturbi psichiatrici e deterioramento cognitivo.
Le cause della malattia di Huntington
La causa della malattia di Huntington è un difetto in un gene (IT15), che codifica per una proteina chiamata huntingtina. I risultati difetto genetico nella produzione di un anormalmente lunga proteine (mutata), che sarebbe responsabile della degenerazione neuronale che si verifica nella malattia. Nel cervello c'è perdita neuronale e depositi huntingtin e altre proteine in forma di inclusioni nucleari e citoplasmatiche a sopravvivere neuroni.
Non vi è alcuna relazione tra la malattia di Huntington e Parkinson, anche se sia in un processo di neurodegenerazione e la deposizione di proteine anomale nel cervello si verifica: hungtintina in HD e sinucleina nel Parkinson. In Parkinson, a differenza di quanto accade in HD, i movimenti del paziente sono caratteristicamente rallentamento. La malattia di Huntington vi è un eccesso di mobilità (ipercinesia).
Vi è anche un importante esposizione clinica tra HD e altre malattie ereditarie neurologiche.
Quando e come appare la malattia di Huntington
L'età media di insorgenza dei sintomi è di 38 anni, con confini variabili tra la seconda e la settima decade di vita. Il quadro clinico set si compone di tre manifestazioni cardine: ipercinesia coreico, disturbi neuropsichiatrici e deterioramento cognitivo. La corea colpisce i muscoli, ma anche le estremità assiali. Inoltre, ci disartria e disfagia volte come risultato di una disfunzione dei muscoli faringei e esofageo superiore. Il quadro dei disturbi neuropsichiatrici è molto variabile, da cambiamenti di personalità sottili e la depressione a disturbi psicotici gravi. Vi è un'alta incidenza di suicidi in famiglie con malattia di Huntington.
La diagnosi differenziale della malattia di Huntington
La diagnosi si basa sui dati clinici e controllare la trasmissione verticale di trasmissione autosomica dominante. Neuroimaging (TC e RM) mostrano alterazioni tipiche. Conferma di EH, e la diagnosi presymptomatic possono essere effettuate con tecniche genetiche molecolari che dimostrano la patologica espansione tripletta CAG nel gene IT15.
Sappiamo che tra il 1 e il 7% dei pazienti clinicamente diagnosticati EH non hanno una mutazione del gene IT15. Questi soggetti con una diagnosi clinica di Huntington 's malattia, ma IT15 negativo, è venuto per essere chiamato malattia di Huntington li-simile (HDL).
Oltre a cause genetiche, ci sono molti Coree acquisiti da considerare nella diagnosi differenziale della corea in HD. Questi includono la corea di Sydenham, malattia di Wilson, ipertiroidismo, malattia coreico discinesia indotta da farmaci (discinesia tardiva) o di Gilles de la Tourette.
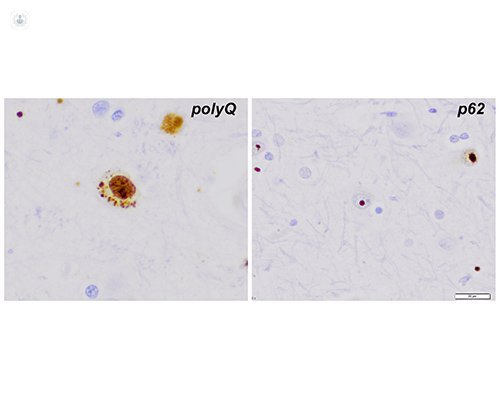
Istologia mostra anche la significativa perdita di neuroni spinosi medi nei gangli basali, l'accumulo intranuclear di ripetizioni CAG, come si è visto dal immunoistochimica con anticorpi contro espansioni polyglutamine (pannello di sinistra, polyQ). inclusioni intranucleari compatte sono visibili utilizzando anticorpi anti - ubiquitina o anti - P62 immunostaining (pannello di destra, p62)
La malattia di Huntington Previsioni
La malattia di Huntington segue invariabilmente un decorso clinico progressivo che è più veloce nelle forme ad esordio giovanile. La durata media di HD è di 19 anni, con i limiti tra i 10 ei 25 anni.
Il trattamento della malattia di Huntington
Il trattamento può alleviare alcune dimostrazioni, ma non ritardare o insorgenza o la progressione dei sintomi. Il trattamento deve essere multidisciplinare e dovrebbe sostenere non solo il paziente, ma anche le loro famiglie, per cui si consiglia di intervenire, oltre ad esperti di neurologia , assistenti sociali, psicologi e altri professionisti che conoscono i problemi di questi pazienti.
Gli unici farmaci che si sono dimostrati efficaci nel controllare la corea comprendono gli antagonisti di recettori della dopamina nello striato di archiviazione o il rilascio di dopamina (reserpina e tetrabenazina) (neurolettici) inibitori. Non esiste un trattamento efficace per la demenza, l'aspetto più debilitante della malattia, ma antipsicotici e antidepressivi sono utili nella terapia di altre manifestazioni neuropsichiatriche.
In aggiunta, la malattia di Huntington è essenziale per una consulenza genetica appropriata. Rilevare portatori della malattia, stadi presintomatici, è possibile grazie alle nuove tecniche di biologia molecolare. Dal momento che l'applicazione di questi non è esente da problemi legali ed etici in molti ospedali dispongono di un comitato in pazienti, medici, avvocati e medici esperti di etica partecipanti, al fine di valutare, caso per caso, se per la loro attuazione.

